
Thalassemia is an inherited blood disorder in which the body makes an unusual form of haemoglobin. Haemoglobin is the protein particle in red platelets that carries oxygen. The condition results in excessive decrease of red blood cells, which prompts anaemia. Individuals with Thalassemia disease are not able to produce enough haemoglobin, which causes severe anaemia. Anaemia is a condition where your body needs more healthy red blood cells. Natural Health talked to Dr. Tengku Ahmad Hidayat, Consultant Clinical Hematologist, Beacon Hospital, in hopes to uncover more about this dreaded condition.
Q: Are there specific types of thalassemia?
Dr Tengku AH: Hemoglobin is made of two proteins, namely Alpha globin and Beta globin.
Thalassemia happens when there is a defect in a gene that helps to regulate one of these proteins’ development. There are also two main types of Thalassemia:
- Alpha thalassemia happens when alpha globin protein-related genes or genes are missing or changed (mutated).
- Beta thalassemia happens when development of the beta globin protein is impaired by similar gene defects.
Q: What are the causes of thalassemia?
Dr Tengku AH: Thalassemia happens when one of the genes involved in the development of haemoglobin has an abnormality or mutation. This genetic abnormality is inherited by one’s parents. In general, individuals with mild to extreme types of thalassemia typically find out about their condition because early in life they show signs of severe anaemia. Individuals with less serious types of thalassemia can find out only because they have symptoms of anaemia or if a doctor discovers anaemia during a regular blood test or a test. You have a better risk of inheriting a more severe form of the disease if both of your parents are carriers of thalassemia.
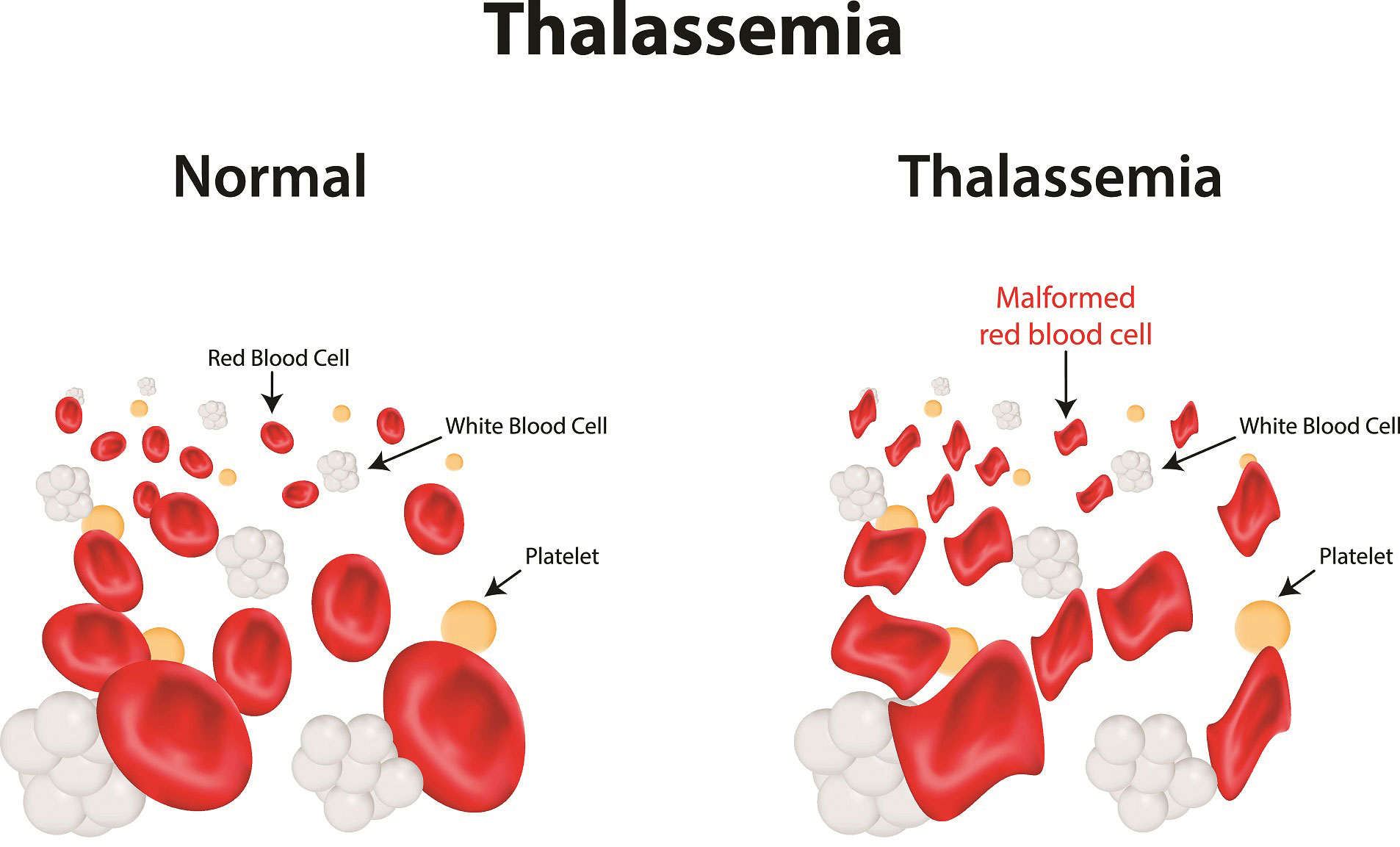
Q: How can thalassemia be detected?
Dr Tengku AH: Blood tests, such as a full blood count (FBC) and special haemoglobin tests, are used to detect thalassemia. A full blood count (FBC) determines the amount of haemoglobin and various types of blood cells, such as red blood cells, in a sample of blood. In the blood of people with thalassemia, there are less healthy red blood cells and less haemoglobin than average. Red blood cells in people with the alpha or beta thalassemia trait can be smaller than average.
Haemoglobin analysis is used to determine which forms of haemoglobin are present in a blood sample. The alpha or beta globin protein chains of haemoglobin are damaged in people with thalassemia.
Q: If I have thalassemia, how does it affect my body? Are there any signs and symptoms of thalassemia?
Dr Tengku AH: The signs of thalassemia vary. Some of the more prevalent ones include infections; increased iron content; deformities of the bones; dark urine; delayed production and growth; excessive fatigue or exhaustion; pale or yellow skin.
Not everybody has symptoms of thalassemia that are apparent. In childhood or adolescence, symptoms of the disease also known as appear to turn up later.
Q: Are there any complications of thalassemia?
Dr Tengku AH: People with moderate to serious thalassemia will now live for longer thanks to improved therapies. As a result, these people must deal with the long-term consequences of their illnesses.
Complications are still common and include heart disease, liver disease, infections and osteoporosis.
Q: Who is particularly at risk of thalassemia?
Dr Tengku AH: Individuals with mild to extreme types of thalassemia have severe anaemia symptoms early in life, they normally find out about their condition in childhood. Individuals with less serious types of thalassemia can only find out when they are experiencing anaemia symptoms, or because a doctor discovers anaemia during a regular blood test or another test. Due to the fact that thalassemia are hereditary, they can run in families. Some individuals learn about their thalassemia from relatives who have the same disease.
Q: Can thalassemia be treated or managed?
Dr Tengku AH: The type of treatment a person requires is determined by the severity of his or her thalassemia. The more severe the thalassemia, the less haemoglobin the body produces, and the greater the risk of anaemia.
Increasing the number of red blood cells in the body to carry oxygen is one way to treat anaemia. A blood transfusion is a safe and normal procedure that involves receiving blood through a small plastic tube inserted into one of your blood vessels.
Since a thalassemia patient’s body produces too little haemoglobin, some people with thalassemia – commonly thalassemia major – require daily blood transfusions. People with thalassemia intermedia (not as bad as major, but not as mild as trait) may need blood transfusions on occasion, such as when they are sick or have an infection.
People with thalassemia minor or trait do not normally need blood transfusions unless they are either anaemic or have a moderate anaemia. A dietary B vitamin known as folic acid is often prescribed to people with thalassemia to help treat anaemia. Folic acid can aid in the development of red blood cells. Folic acid therapy is commonly used in conjunction with other treatments.
Q: Can thalassemia be prevented?
Dr Tengku AH: Since thalassemia is hereditary, there is no way to avoid them (passed from parents to children through genes). Prenatal tests, on the other hand, can detect these blood disorders before birth. Family genetic studies can aid in determining whether people have thalassemia-causing haemoglobin genes that are missing or altered. If you have thalassemia in your family and are considering having children, consult your doctor and a genetic counsellor. They can help you figure out whether you’re at risk of passing the disease on to your children.
Q: What is the possible care that thalassemia patients need?
Dr Tengku AH: For people with mild to extreme thalassemia, survival and quality of life have increased because of the following reasons:
- Blood transfusions are also available to a larger number of people.
- The number of infections caused by blood transfusions has decreased as a result of blood screening. Treatments for other types of infections have also progressed.
- There are iron chelation therapies available that are more convenient for certain people.
- Blood and marrow stem cell transplants have healed some patients.
Living with thalassemia can be difficult, but there are many ways to cope such as:
- Follow up with your treatment plan: It is important that you adhere to your doctor’s treatment plan. For instance, get blood transfusions as directed by your doctor, and take your iron chelation medication as directed. Chelation therapies are also available in a variety of forms, including injections and tablets. Your doctor will discuss which care option is best for you.
- Get Consistent Medical Care: Keep all of your doctor’s appointments and get any tests that your doctor suggests. These assessments can include the following:
- Every three months, full blood counts and blood iron levels tests are performed.
- Heart function, liver function, and viral infection tests are done once a year
- Tests every year to see if your liver has built up any iron.
- Blood transfusions must be checked on a regular basis to ensure that they are functioning properly.
- Additional tests as required and advised by your Haematologist.
Q: How do blood transfusions affect my body?
Dr Tengku AH: Iron overload can occur in people who receive a lot of blood transfusions. Since red blood cells contain a lot of iron, the iron from all of the transfusions will accumulate in the body over time. When iron builds up in organs like the heart, liver, and brain, it may make it difficult for these organs to function properly. People with thalassemia can need chelation therapy, which involves doctors administering a medication – either a pill or a shot under the skin – to extract excess iron from the body until it accumulates in the organs.
When a person receives a blood transfusion, their risk of developing a condition known as “alloimmunization” increases. When a person’s immune system perceives blood from a transfusion as toxic and attempts to kill it, this is known as alloimmunization. All immunized people can still have blood transfusions, but the blood must be screened and compared to their own blood to ensure that it won’t be destroyed by their immune system. This takes time, and people with alloimmunization may have to wait longer for blood or have a more difficult time finding blood that won’t be destroyed by their bodies.
Another problem for people who receive a lot of blood transfusions is the blood’s protection. Some diseases, such as hepatitis, may be transmitted by blood. There is a very small chance of contracting an infection after receiving a blood transfusion.
Q: Is blood transfusion required for all the types of Thalassemia?
Dr Tengku AH: Since thalassemia patients produce too little haemoglobin, some people with thalassemia – commonly thalassemia major – require daily blood transfusions. Individuals with thalassemia intermedia (not as extreme as major, but not as mild as trait) would need blood transfusions on occasion, such as when they are ill or get an infection. Individuals with thalassemia minor or trait do not normally need blood transfusions.
Q: What are the things that a Thalassemia Carrier should know or to be aware about?
Dr Tengku AH: An individual who carries the thalassemia trait is healthy. Being a thalassemia carrier has no documented health consequences that necessitate medical attention. The thalassemia trait will not affect the ability to work, eat, or exercise.
Carriers with thalassemia have fewer red blood cells, which may induce a slight anaemia.
As for family planning, if you’re intending to have a new-born and know you’re a carrier, having your partner checked is a wise decision. If you’re both carriers and intending to have a child, consult a haematologist to learn about the risks to your children and what your options are.
Women that have the gene may have a higher risk of anaemia during pregnancy. It’s important to speak with a Haematologist about the symptoms.
Conclusively, the treatment for thalassemia has progressed over time. People with thalassemia, whether mild/intermediate or major, are now living longer and enjoying a higher quality of life. Complications from thalassemia and their treatments, on the other hand, are common. People with moderate to extreme thalassemia must adhere to their care plans religiously. To stay as safe as possible, they must take care of themselves.
 Q & A with Dr. Tengku Ahmad Hidayat
Q & A with Dr. Tengku Ahmad Hidayat
Consultant Clinical Haemotalogist,
Beacon Hospital